 SpanishSpanish
SpanishSpanishCitar los tipos específicos de malformaciones, ayudando a comprender el tema
¿Ha oído hablar alguna vez del síndrome de Apert? Aunque poco conocido, este síndrome genético afecta a uno de cada 100.000 a 160.000 nacidos vivos.
El síndrome de Apert es una malformación congénita grave que implica la unión precoz de los huesos del cráneo y de la base del cráneo (craneofacioestenosis).
Por lo tanto, este síndrome es responsable de causar un desarrollo anormal de la estructura craneal y facial de una persona. Como resultado, los bebés con esta malformación nacen con una forma de cabeza y una cara diferentes.
El Síndrome de Apert sólo se da en aquellas personas que tienen una mutación en uno de los genes responsables de los fibroblastos, que son los encargados de promover la unión de los huesos del cráneo mientras el bebé aún se está desarrollando en el útero materno. Como consecuencia, los huesos de la calota y de la base del cráneo se unen de forma precoz e incorrecta, lo que puede poner en peligro la estética del cráneo, la cara y también el desarrollo del cerebro del niño.
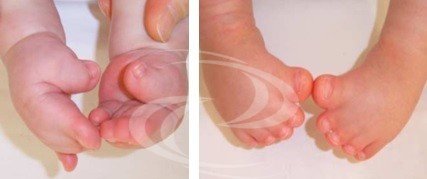
Además, los bebés Apert también nacen con articulaciones de los dedos de manos y pies que parecen estar "pegadas", lo que también se conoce como sindactilia compleja. Esta sindactilia múltiple de los dedos es una característica definitoria del síndrome y ayuda a realizar el diagnóstico y a diferenciarlo de otros síndromes craneofacioestenosis, como el síndrome de Crouzon y el síndrome de Pfeiffer.
Debido a la unión prematura de los huesos del cráneo, el cerebro queda muy constreñido y su crecimiento y desarrollo pueden verse perjudicados. Por otra parte, la unión de los huesos de la base del cráneo impide el crecimiento normal de la cara. Como resultado, los afectados por el síndrome de Apert presentan algunas características físicas reconocibles:
Además, los afectados por el síndrome pueden presentar los siguientes síntomas: apnea del sueño (trastorno en el que la persona deja de respirar durante unos segundos mientras duerme), problemas de audición e infecciones de oído sucesivas y ataques constantes de sinusitis.
El síndrome de Apert puede desarrollarse por dos motivos:
Por sus características físicas, el síndrome de Apert puede diagnosticarse nada más nacer el bebé o mediante pruebas genéticas para confirmar la existencia de la mutación.
Aunque no existe cura para el síndrome, es posible tratar esta malformación con cirugía plástica. Estos procedimientos quirúrgicos se encargan de corregir las alteraciones del cráneo del bebé y los casos de sindactilia. Además, la cirugía permite que el cerebro del recién nacido se desarrolle y evita daños mentales.
Para que el tratamiento sea más eficaz, la cirugía para corregir los signos craneofaciales del síndrome de Apert se divide en tres partes:
Es importante saber que aunque el tratamiento craneofacial se divide en tres cirugías, esto no significa que no vaya a necesitar otras operaciones a lo largo del proceso, ya que algunos casos pueden ser más graves que otros.
En cuanto al tratamiento de la sindactilia de manos y pies, es posible separar los 10 dedos de manos y pies en la gran mayoría de los casos. Esta parte del tratamiento puede dividirse en dos fases quirúrgicas, que pueden iniciarse en el primer año de vida.
Además de los procedimientos quirúrgicos, otras formas de tratar el síndrome de Apert incluyen tratamientos para mejorar las condiciones de vida del enfermo, por ejemplo: medicación para tratar la sinusitis y la otitis, tratamiento intensivo para la apnea del sueño y colirios diarios para evitar la sequedad ocular, entre otros tratamientos que mejoran los síntomas del síndrome.

Síndrome descrito por Crouzon en 1912, combina craneostenosis e hipoplasia del tercio medio facial. En la mayoría de los casos, la craneoestenosis afecta a las dos suturas coronales.
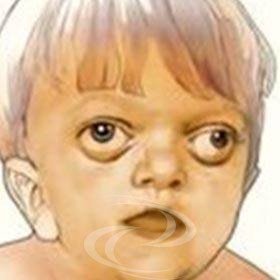
Las alteraciones faciales son características: hiperteleorbitismo (las órbitas se separan), exorbitismo (con órbitas poco profundas, lo que provoca el efecto de "ojos saltones") y retrusión de todo el tercio medio de la cara. Suele pasar desapercibida al nacer, pero tiende a hacerse más evidente en torno a los 2 años y empeora progresivamente.
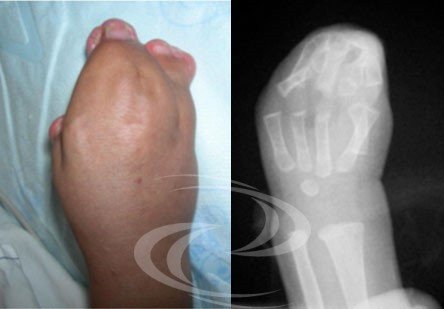
Síndrome descrito por Rudolf Pfeiffer en 1964, presenta una braquicefalia (cierre de las suturas craneales coronales) asociada a una sindactilia membranosa variable de las manos y los pies y, sobre todo, a un agrandamiento de los pulgares y los dedos gordos ("dedo gordo agrandado"), con una marcada desviación hacia el interior. A menudo se describe braquidactilia, estenosis de codo e incluso sinfalangia.
Manos y pies en el síndrome de Pfeiffer, con sindactilia membranosa variable y el característico agrandamiento y desviación de los pulgares y los dedos gordos de los pies.
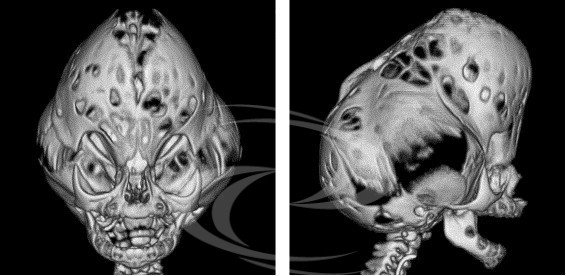
Se describen formas graves con cráneo en forma de hoja de trébol. En estos casos, la dismorfia craneal es muy importante, debido al bombeo considerable de las fosas temporales y a la estenosis lateral de las regiones frontoparietales, lo que conduce a un aspecto trilobulado visto de frente. La hidrocefalia congénita es constante. Esta importante dismorfia, a menudo denominada síndrome del cráneo en trébol, también puede observarse en los síndromes de Apert y en la forma precoz del síndrome de Crouzon.
Cráneo con alteración característica en forma de trébol.
El síndrome de Pfeiffer es poco frecuente, con una incidencia aproximada de 1 por 100.000 individuos. La craneoestenosis se produce en asociación con pulgares y dedos gordos cortos y desviados hacia dentro. La fusión prematura de las suturas coronal y lambdoidea puede ir acompañada ocasionalmente de la fusión también de la sutura sagital, lo que da lugar a una forma anormal del cráneo. Se produce un aspecto facial característico, con ensanchamiento de la cabeza y aplanamiento del occipucio, alargamiento de la región frontal, hipoplasia (menor desarrollo) del tercio medio de la cara, nariz corta con dorso nasal bajo y los ojos separados (hipertelorismo ocular). Los pacientes suelen tener los ojos prominentes (proptosis ocular) porque las órbitas son demasiado poco profundas.
Los pulgares y los dedos gordos de los pies son cortos y torcidos, con una desviación típica. Los dedos segundo y tercero están unidos (sindactilia). Otras anomalías pueden ser: retraso mental de grado variable, estenosis del acueducto cerebral con la consiguiente hidrocefalia, implantación baja de las orejas, estenosis (estrechamiento) del conducto auditivo externo, infecciones de oído recurrentes y, con menor frecuencia, hidronefrosis, riñones pélvicos, vejiga subdesarrollada (hipoplásica).
Los pacientes con síndrome de Pfeiffer pueden presentar obstrucción de las vías respiratorias superiores debido a la retrusión del tercio medio de la cara.
Según sus características clínicas, el síndrome de Pfeiffer puede dividirse en 3 subtipos:
Tipo 1: Síndrome de Pfeiffer "clásico", con manifestaciones leves que incluyen braquicefalia, hipoplasia del tercio medio de la cara y anomalías de los dedos de manos y pies. Se asocia a un desarrollo neurológico e intelectual normal y suele tener un buen pronóstico.
Tipo 2: se caracteriza por una deformidad craneal trilobulada, con una prominencia en la parte superior y otra a cada lado de la cabeza, dando un aspecto de "hoja de trébol" (también llamada Kleeblattschädel), aunque esta característica no es exclusiva del síndrome de Pfeiffer y puede darse en otros síndromes o incluso de forma aislada. También presenta proptosis grave (ojos saltones), anomalías en los dedos de manos y pies, anquilosis (articulación) o estenosis del codo, retraso en el desarrollo mental y complicaciones neurológicas. El cráneo en forma de trébol puede limitar el crecimiento del cerebro y la proptosis grave puede causar una pérdida visual importante.
Tipo 3: similar al tipo 2, pero sin deformidad craneal en forma de trébol. La ausencia de cráneo en forma de trébol puede dificultar el diagnóstico. Los tipos 2 y 3 ocurren esporádicamente y conllevan un mayor riesgo de muerte prematura debido a graves alteraciones neurológicas y problemas respiratorios. Puede producirse un solapamiento clínico entre los tres tipos.
Diagnóstico del síndrome de Pfeiffer
El diagnóstico suele ser clínico, basado en la presencia de craneoestenosis y alteraciones en los pulgares y los dedos de los pies. Genéticamente, se producen mutaciones en los genes receptores del factor de crecimiento de fibroblastos (FGFR1 y FGFR2). Estos genes actúan en la señalización de las células en respuesta a su entorno, regulando los procesos de proliferación, diferenciación y migración celular. Una mutación en uno de estos genes provoca una señalización prolongada, que puede dar lugar a una maduración precoz de las células óseas y a una fusión prematura de los huesos del cráneo, las manos y los pies.
Se trata de un diagnóstico diferencial con otros síndromes de craneofacioestenosis, como los síndromes de Crouzon y Apert, tratados en otro lugar. De hecho, las similitudes clínicas son grandes, especialmente en lo que respecta al aspecto orbitario (órbitas poco profundas con proptosis ocular) y la retrusión del tercio medio de la cara, aunque genéticamente son distintos.
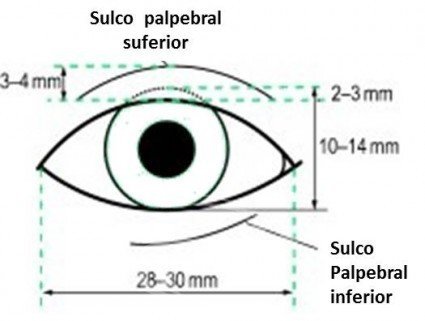
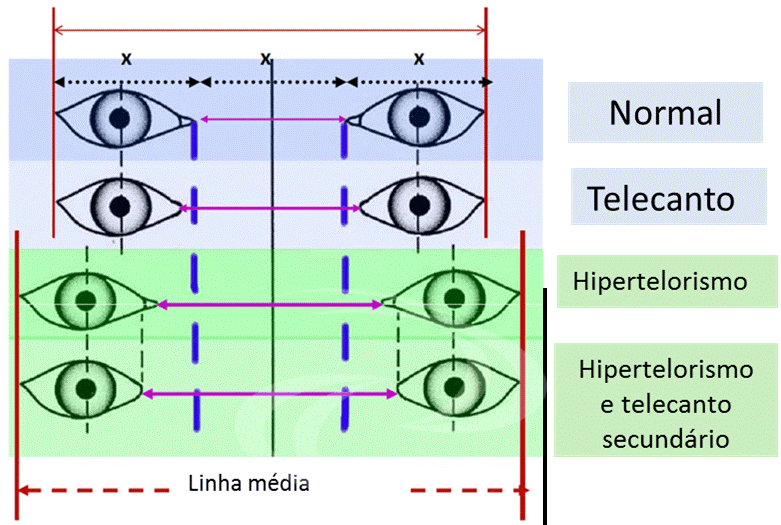
El término hipertelorismo orbitario o hiperteleorbitismo, o simplemente hipertelorismo, corresponde a un aumento de la distancia entre las órbitas, o aumento de la distancia interorbitaria. La distancia entre las órbitas puede obtenerse midiendo la distancia entre los dacriones, o el punto donde convergen las suturas frontal, nasal y lagrimal en la cresta lagrimal anterior, es decir, la distancia entre los márgenes orbitarios medial o interno.
Puede producirse un aumento de la distancia entre los ángulos medios de los ojos sin que exista un verdadero hipertelorismo, como ocurre en los traumatismos faciales en los que se produce un telecanto traumático. La distancia entre las pupilas tampoco significa hipertelorismo, ya que en el estrabismo divergente aumenta la distancia interpupilar, sin que exista un aumento de la distancia entre las órbitas.
La distancia normal entre las órbitas varía en función de la edad, la raza y el sexo masculino o femenino del paciente. En general, la medida estándar para los adultos es la distancia interorbitaria de 30 mm.
Clasificación del hipertelorismo
El hipertelorismo puede clasificarse en leve, moderado o grave a medida que aumenta la distancia entre las órbitas:
Peso ligero: entre 30 y 34 mm
Moderado: >34 y <40mm
Grave: más de 40 mm
Causas del hipertelorismo
El hipertelorismo orbitario está relacionado con malformaciones congénitas, ya sean debidas a factores genéticos o extrínsecos. Las principales causas de hipertelorismo son: hendiduras faciales poco frecuentes, como la hendidura de Tessier 14, displasia frontonasal, displasia craneofrontonasal (componente genético), encefaloceles frontales, encefaloceles nasoetmoidales, síndromes de Apert y Crouzon.
Algunos problemas estéticos pueden acompañar al hipertelorismo, como el acortamiento de la nariz, la hendidura alar, la punta nasal bífida, la frente malformada, la implantación anormal de la línea del cabello, el espaciado de las cejas, la forma anormal del ojo, la malposición de la órbita, la distopía del ángulo lateral del ojo, el acortamiento o la hipoplasia de la mejilla, la retrusión o la hipoplasia del maxilar, la obstrucción o el ensanchamiento del conducto lagrimal, la hipertrofia del tejido subcutáneo de la zona orbito-nasal.


Dra. Clarice Abreu
CRM 52.77806-0
Hola, ¿desea concertar una cita en persona o en línea?

