


¿Ha oído hablar alguna vez del síndrome de Apert? Aunque poco conocido, este síndrome genético afecta a uno de cada 100.000 a 160.000 nacidos vivos.

¿Qué es el síndrome de Apert?
A síndrome de Apert é uma malformação congênita grave, que associa uma união precoce dos ossos do crânio e da base do crânio (craniofacioestenose). Essa síndrome é responsável, portanto, por causar o desenvolvimento anormal da estrutura craniana e facial de uma pessoa. Por isso, os bebês portadores dessa malformação nascem com o formato da cabeça e a face diferentes.
A Síndrome de Apert só acontece naqueles que possuem uma mutação em um dos genes responsáveis pelos fibroblastos, que são encarregados de promover a união dos ossos do crânio ainda durante o desenvolvimento do bebê no útero da mãe. Com isso, há uma união precoce e errônea dos ossos da calota craniana e da base do crânio, o que pode prejudicar a estética do crânio, da face e também o desenvolvimento do cérebro da criança.
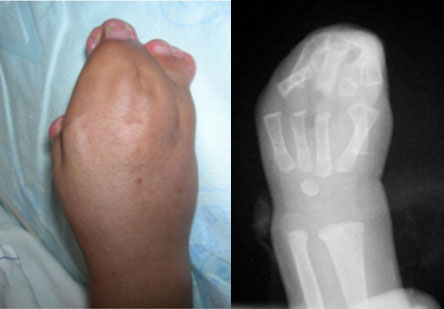
Além disso, os bebês portadores de Apert também nascem com junções dos dedos das mãos e dos pés que parecem estar todos “colados”, o que também é conhecido como sindactilia complexa. Essa sindactilia de múltiplos dedos é uma característica definidora da síndrome e ajuda a fazer o diagnóstico e a diferenciar de outras síndromes de craniofacioestenoses, como a Síndrome de Crouzon e a Síndrome de Pfeiffer.
¿Cuáles son los síntomas del síndrome?
Devido à junção prematura dos ossos do crânio, o cérebro fica muito constrito e pode ter o seu crescimento e desenvolvimento prejudicados. A junção dos ossos da base do crânio, por outro lado, impede que a face cresça normalmente. Com isso, os portadores da Síndrome de Apert terão algumas características físicas reconhecíveis, são elas:
– Face afundada ou retraída;
– Olhos distantes um do outro, devido ao crescimento anormal do crânio;
– Uma testa longa e, consequentemente, uma cabeça de formato mais longo do que o normal.
Além disso, é possível que o portador da síndrome possua os seguintes sintomas: apnéia do sono (distúrbio em que a pessoa pára de respirar por alguns segundos enquanto está dormindo), problemas de audição e sucessivas infecções no ouvido e crises constantes de sinusite.
Causas del síndrome de Apert
El síndrome de Apert puede desarrollarse por dos motivos:
1 – Um dos progenitores possui o gene
Nesse caso, um dos pais possui em seu DNA o gene que causa a síndrome. Dessa forma, como o bebê carrega metade dos genes do pai e a outra metade da mãe, esse pode ser um dos motivos que leva ao desenvolvimento dessa malformação.
2 – Mutação genética desconhecida
Assim como diz o nome, pode acontecer casos em que nenhum dos pais possuem a carga genética da síndrome, mas, mesmo assim, acontece uma mutação durante a gestação e o bebê desenvolve o Apert. Mesmo após mais de um século de sua descoberta, ainda não foi possível saber qual é a razão da mutação. Entretanto, alguns estudiosos sobre o assunto afirmam que gestação em idade avançada pode desencadear esse acidente genético.
Por sus características físicas, el síndrome de Apert puede diagnosticarse nada más nacer el bebé o mediante pruebas genéticas para confirmar la existencia de la mutación.
¿Cómo tratar el síndrome de Apert?
Aunque no existe cura para el síndrome, es posible tratar esta malformación con cirugía plástica. Estos procedimientos quirúrgicos se encargan de corregir las alteraciones del cráneo del bebé y los casos de sindactilia. Además, la cirugía permite que el cerebro del recién nacido se desarrolle y evita daños mentales.
Para melhor eficácia do tratamento, as cirurgias para correção dos sinais craniofaciais da Síndrome de Apert são divididas em três partes:
1 – Cirurgia para remodelamento craniano: feita nos primeiros meses de vida da criança, essa primeira cirurgia tem como objetivo separar as partes que foram fundidas precocemente no crânio, como uma tentativa de expandir o espaço intracraniano e permitir que o cérebro cresça e se desenvolva normalmente.
2 – Cirurgia para avanço da face: feita em torno dos 6-8 anos de idade, a cirurgia consiste em “corrigir” os ossos da face, visto que, conforme a criança se desenvolve, a face se mantém retraída e não cresce normalmente. Com isso, o cirurgião responsável tentará colocar os ossos da região do nariz e maxila na posição em que deveriam estar.
3 – Cirurgia para correção da distância aumentada entre os olhos (hipertelorismo): a terceira e última cirurgia é feita para corrigir a região dos olhos, diminuindo o espaço entre eles.
Es importante saber que aunque el tratamiento craneofacial se divide en tres cirugías, esto no significa que no vaya a necesitar otras operaciones a lo largo del proceso, ya que algunos casos pueden ser más graves que otros.

En cuanto al tratamiento de la sindactilia de manos y pies, es posible separar los 10 dedos de manos y pies en la gran mayoría de los casos. Esta parte del tratamiento puede dividirse en dos fases quirúrgicas, que pueden iniciarse en el primer año de vida.
Além dos procedimentos cirúrgicos, outro jeito de tratar a Síndrome de Apert inclui tratamentos para melhorar a condição de vida do portador, por exemplo: medicação para tratar sinusites e otites, tratamento intensivo para apneia do sono e utilização de colírios diariamente para prevenir secura nos olhos, entre outros tratamentos que melhorem os sintomas da síndrome.
4 dúvidas sobre a Síndrome de Apert

1 – Qual é a expectativa de vida para os portadores da Síndrome de Apert
A expectativa de vida para uma pessoa que nasceu com a síndrome pode variar. Tudo depende do quanto a anomalia afetou o desenvolvimento do bebê. Por exemplo, uma criança que conseguiu sobreviver aos primeiros anos de vida e não teve mais nenhum tipo de complicação, pode ter uma expectativa de vida igual à de uma que nasceu sem Apert.
2 – Se eu tiver essa síndrome, meu filho também terá?
Por ser uma síndrome genética, caso um dos progenitores tiver o gene, existe 50% de chances do seu filho também nascer com essa malformação craniofacial.
3 – Caso eu tenha, existe alguma forma de evitar que meu filho nasça com a síndrome?
Se você ou seu parceiro (a) apresentam o gene da síndrome, o mais indicado é que utilizar o método de fertilização in vitro para gerar um bebê. Dessa forma, será possível selecionar o embrião que não possui nenhum sinal de Apert.
Entretanto, é importante saber que, mesmo que o embrião inicialmente não tenha o gene, é possível que a mutação se desenvolva durante a gestação.
4 – Meu filho terá um desenvolvimento mental normal?
Para que portadores da Síndrome de Apert tenham um desenvolvimento normal, é preciso que a cirurgia de tratamento craniano seja feita o mais rápido possível para evitar que o pouco espaço ou a malformação afetem o crescimento do cérebro. Com as cirurgias corretas e com acompanhamento médico, é muito provável que o bebê consiga se desenvolver normalmente.
É preciso lembrar o quão importante é a informação para mães e pais de bebês com Síndrome de Apert. Sem conhecimento, será muito mais complicado lidar com o diagnóstico. Por isso, caso você conheça alguém que possua essa síndrome ou tem um filho que seja portador de Apert, indique este texto. 🙂
Veja também: 7 famosos que nasceram com malformações e você não sabia