Síndrome descrita por Apert em 1906, é uma malformação congênita grave, associando uma craniofacioestenose com sindactilias (junções) ósseas e membranosas das quatro extremidades (mãos e pés). A cranioestenose (fusão das suturas cranianas) é sempre bicoronal e geralmente respeita o sistema longitudinal (suturas metópica e sagital), que é extraordinariamente grande mesmo nos primeiros meses de vida. O maxilar superior é muito hipoplásico (pouco desenvolvido), com uma inversão do articulado dentário, e a face é larga, com um nariz em bico de papagaio, hiperteleorbitismo (afastamento das órbitas) constante e um exorbitismo (órbitas rasas, com “olhos saltados”) que pode ser grave. A sindactilia (junção) dos dedos das mãos e dos pés é total, respeitando muitas vezes o polegar e/ou o quinto dedo (dando um aspecto em “bloco” das extremidades).
Causas da Síndrome de Apert
A Síndrome de Apert é causada pela mutação (mudança) em um gene que codifica o receptor 2 do Fator de Crescimento de Fibroblasto (do inglês: FGFR 2), localizado no cromossomo 10q26. Duas mutações adjacentes (P253R e S252W) nesse cromossomo podem produzir a Síndrome de Apert, de modo que todos os pacientes investigados apresentam uma ou outra dessas mutações. A causa dessas mutações permanece desconhecida, e cada uma leva a um “tipo” de apresentação específica da síndrome. A mutação P253R estaria associada a uma sindactilia mais grave. Ao contrário, a fenda palatina, é mais frequentemente associada à mutação S252W.
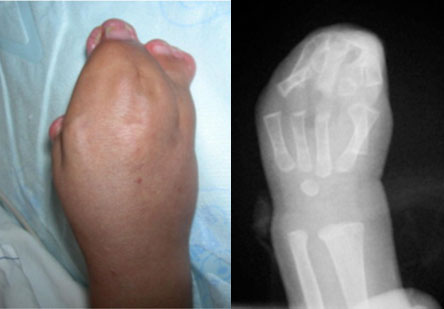
Em geral, tanto a mãe quanto o pai de um bebê com Síndrome de Apert apresentam genes normais. O risco de pais normais terem um bebê acometido é estimado em 1:120.000 e a incidência da síndrome é estimada em torno de 1:65.000 nascimentos. Quando um dos pais é portador da síndrome, o risco de transmissão é de 50%, pois o gene tem um padrão de transmissão autossômica dominante.
Características clínicas
A Síndrome de Apert afeta várias regiões do corpo. Alguns pacientes apresentam várias características, com acometimento mais grave, enquanto outros apresentam quadros mais leves. Entre as principais características, destacamos:
Crânio e face
Como dito anteriormente, a Síndrome de Apert representa uma craniofacioestenose. Isso significa que ocorre uma estenose (fusão) precoce das suturas do crânio e das suturas da base do crânio que afetam o crescimento facial. No crânio, as suturas acometidas são as coronais que correm transversalmente (“de orelha a orelha”). Quando essas suturas estão fechadas, o crescimento cerebral não consegue promover o deslocamento anterior e posterior do crânio, que acaba crescendo apenas para cima. A medida que o bebê cresce, a fronte (testa) tende a se alongar verticalmente. Em geral, a fontanela anterior (“moleira”) se encontra amplamente aberta, juntamente com a sutura sagital e a metópica.
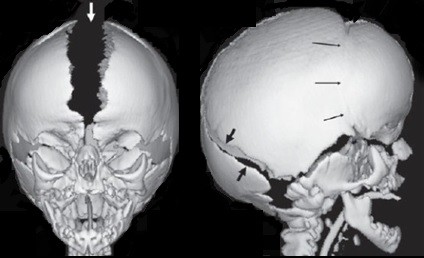
Além das estenoses cranianas, ocorrem ainda estenoses em suturas da base do crânio, que acabam por impedir o crescimento facial adequado. Dessa forma, o terço médio da face (entre os olhos e a maxila) não cresce no sentido ântero-posterior, caracterizando o aspecto clínico de retrusão da face, com órbitas rasas e desproporção entre a maxila e a mandíbula.
Mãos e pés
A característica clínica definidora da Síndrome de Apert é a sindactilia (junção) de todos os dedos das mãos e dos pés, também chamada polissindactilia. Em alguns casos, pode haver a separação entre o polegar e o dedo indicador e mais raramente, entre o dedo anelar e o dedo mínimo.
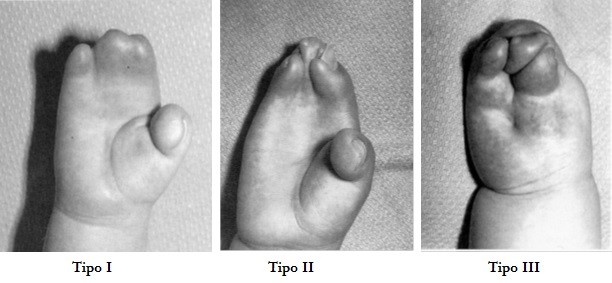
A mão na Síndrome de Apert pode ser classificada em tipos I, II e III de acordo com o número de dedos que estão unidos. Em casos mais leves, apenas o indicador, o dedo médio e o dedo anelar estão unidos. Ocasionalmente, o polegar está completamente sepultado pela pele da palma da mão. Clinicamente, os três tipos diferentes de malformação das mãos são: a mão espalmada, em “pá” (tipo I), a mão constricta em colher, ou mãos em “luva de boxe” (tipo II), e a mão coalescente em forma de “botão de rosa” (tipo III).
Os ossos dos dedos tendem a crescer de forma curva (clinodactilia), de modo que a ponta do polegar cresce desviada para fora e longe do dedo indicador. Além do desvio, o polegar ainda tende a ser mais curto que o normal, caracterizando uma braquiclinodactilia. Os ossos dos demais dedos podem ser mais curtos pela ausência de suas a articulações médias e também podem estar fundidos, normalmente nas falanges distais (simbraquifalangismo). As placas ungueais (unhas) também estão unidas.
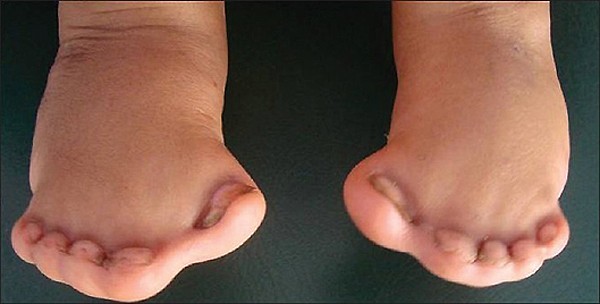
Os dedos dos pés apresentam quase sempre unhas separadas. Fusões ósseas entre os dedos dos pés são raras e quando acontecem são apenas entre alguns dedos. O dedão do pé (hálux) é mais curto que o normal e, como os polegares, crescem desviados para longe do pé. Esse alargamento do pé pode tornar difícil a adaptação de sapatos e as famílias acabam comprando sapatos muito maiores que o tamanho do pé para poder acomodar a largura dos dedos.
Olhos
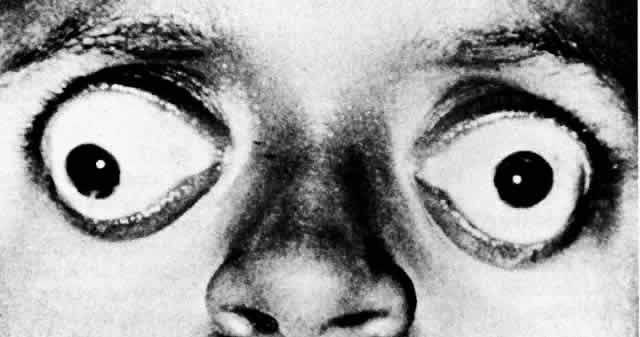
Crianças com Síndrome de Apert parecem ter olhos grandes, ou “olhos saltados”. Na verdade, o tamanho dos olhos é normal nessas crianças, porém o volume das órbitas está diminuído. Trata-se, portanto, de uma desproporção continente-conteúdo, em que as órbitas rasas não envolvem completamente o volume ocular normal.
Alguns pacientes apresentam essa desproporção de forma tão grave que os olhos podem eventualmente luxar (se deslocar anteriormente para fora da órbita), devendo ser reposicionados rapidamente.

A falta de cobertura palpebral adequada pode levar ainda à exposição constante da córnea, podendo levar a ulcerações corneanas e consequente perda visual.
A diminuição da acuidade visual pode também decorrer de estados de hipertensão intracraniana prolongados, que podem levar a atrofia do nervo óptico.
Frequentemente, os pacientes com Síndrome de Apert apresentam algum grau de estrabismo (desalinhamento dos olhos, que apontam para direções diferentes), devido a um desequilíbrio entre os músculos responsáveis pela movimentação dos olhos. Esse desequilíbrio deve ser corrigido para evitar que se intale a ambliopia, que corresponde à diminuição da acuidade visual de um ou ambos os olhos.
Pele
Pacientes com Síndrome de Apert tendem a ter a pele mais oleosa. Alguns pacientes podem apresentar sudorese excessiva, também conhecida como hiperhidrose, que resulta de uma atividade excessiva das glândulas sudoríparas, responsáveis pela produção do suor. Na adolescência, é comum o desenvolvimento de acne.
Orelhas
As orelhas geralmente apresentam forma e contornos normais, porém são posicionadas um pouco abaixo do normal. O ouvido interno pode estar acometido como resultado da retrusão da face, ou mesmo da presença de fenda palatina, que prejudica a drenagem interna normal do conduto auditivo. Dessa forma, as crianças com Síndrome de Apert podem desenvolver otites de repetição. Nesses casos, pode estar indicado o uso de tubos de ventilação (“carretel”) para diminuir o desenvolvimento de infecções de repetição.
Boca, palato e via aérea
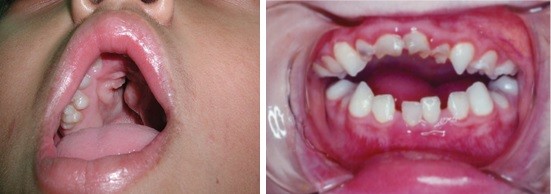
Como resultado do encurtamento da face, o palato (“céu da boca”) faz um arco bem alto. Esse arco empurra o assoalho do nariz, tornando a respiração pelo nariz muito difícil. O palato frequentemente está fendido (em aproximadamente 30% dos casos). Tipicamente, a fenda ocupa apenas o palato posterior (palato secundário) e raramente se extende para a região anterior. Às vezes, o palato é tão alto (palato ogival) e estreito que forma uma pseudo fenda palatina, embora esteja completamente fechado.
A retrusão facial com hipoplasia da maxila leva a uma inversão do articulado dentário, além de toque prematuro dos molares com mordida aberta anterior.
A via aérea é quase sempre comprometida. Os pacientes apresentam respiração ruidosa, especialmente à noite. Isso ocorre porque com a face retrusa, o nariz também fica reduzido de tamanho e portanto pouco ar pode passar. O teto da boca é mais baixo na região posterior, quase tocando a língua. Algumas vezes, a língua cai e oclui a passagem de ar. Além disso, a traquéia pode ser um pouco estreitada, contribuindo ainda mais para a dificuldade respiratória. Uma polissonografia (exame que estuda a respiração durante o sono) deve ser realizada regularmente para medir a quantidade de oxigênio no sangue e qual o grau de apnéia do sono que se encontra o paciente em estudo.
Cérebro
Muitas crianças com Síndrome de Apert apresentam dilatação dos ventrículos cerebrais. O simples aumento do volume dos ventrículos não indica necessidade de tratamento, porém quando essa dilatação ventricular é acompanhada de um aumento na pressão intracraniana, pode ser indicada a colocação de uma válvula que faz a drenagem do excesso de líquido cérebro espinhal (liquor) para a cavidade abdominal. Também conhecida como derivação ventrículo-peritoneal, a válvula quando colocada em geral permanece pelo resto da vida.
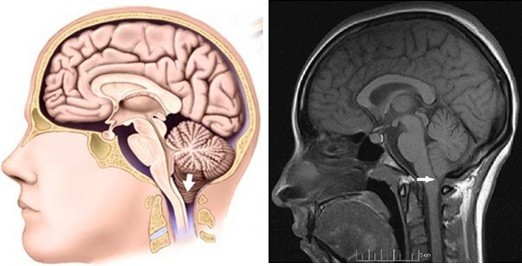
Outra condição que pode ocorrer é chamada de herniação da tonsila cerebelar, conhecida como Malformação de Chiari. Sua ocorrência é incomum na Síndrome de Apert, no entanto, é muito importante investigar essa condição. A base do crânio tem um orifício chamado forame magno, por onde passa o cordão espinhal. Algumas vezes, parte do cérebro chamada de tonsila cerebelar pode ser empurrada para baixo dentro desse orifício como uma rolha em uma garrafa de vinho. Se isso acontece, pode haver diminuição do fluxo de líquido cerebro espinhal e a criança pode desenvolver um tipo específico de apnéia do sono chamado apnéia central. O ideal seria investigar anualmente o desenvolvimento dessa condição, que pode refletir um aumento na pressão intracraniana, indicando a necessidade de avançar a fronte ainda mais para ganhar mais espaço. Trata-se de condição clínica melhor avaliada com Ressonância Magnética.
Outras alterações cerebrais que podem ser identificadas em exames de imagem são agenesia (ausência) do corpo caloso e septo pelúcido, entre outras alterações estruturais menos frequentes.
O aumento da pressão intracraniana é uma preocupação constante na Síndrome de Apert. Crianças com Síndrome de Apert apresentam desproporção entre o crescimento do cérebro e o crescimento do crânio. Como as suturas cranianas estão fechadas (cranioestenose), o crânio não acompanha o crescimento cerebral, podendo levar a compressão do cérebro e o consequente aumento da pressão intra-craniana. Essa condição deve ser acompanhada por um médico especializado, para que o tratamento cirúrgico de expansão craniana seja indicado em momento oportuno.
Muitas crianças com Síndrome de Apert apresentam atrasos do desenvolvimento. Entre os motivos para esses atrasos, além de alterações estruturais do sistema nervoso central, destacamos a apnéia do sono, com graus de hipóxia (baixa oxigenação cerebral) variáveis e crônicos, além da hipertensão intracraniana.


